#10 Dr. Aubrey de Grey and Dr. Rhonda Patrick Talk Aging
This episode is available in a convenient podcast format.
These episodes make great companion listening for a long drive.

The BDNF Protocol Guide
An essential checklist for cognitive longevity — filled with specific exercise, heat stress, and omega-3 protocols for boosting BDNF. Enter your email, and we'll deliver it straight to your inbox.
Dr. Aubrey de Grey is a biomedical gerontologist and the founder of the SENS Research Foundation which aims to find technologies that can repair the various types of damage that occur during the aging process.
"CRISPR I would single out as a particularly important advance because there are definitely quite a few things we're going to have to do in getting this damage repair to be comprehensive that involve genetic modifications." - Dr. Aubrey de Grey Click To Tweet
Learn more about Dr. Aubrey De Grey
- SENS Research Foundation
- @aubreydegrey on Twitter
- @senstweet on Twitter
- Aubrey de Grey on Facebook
- SENS Research Foundation on Facebook
- Wikipedia Profile on Dr. Aubrey de Grey
People mentioned
-
What is aging and how is it characterized.
-
The epigenetics of our cells change as we age in order to accomodate aging effects.
-
-
Crispr/Cas9 has potential to change our genetics to curb the aging process.
-
-
-
Inflammation may be a double-edged sword that can protect you from infection but promote diseases of aging such as atherosclerosis.
-
Parabiosis is the physiological joining of two organisms and is being researched on for its potential to slow aging.
-
The effect of anti-aging remedies differs depending on a person's current health status.
-
Despite the USA being #47 in life expectancy, it is only 5 years different from Japan #1.
-
How does Crispr/Cas9 work and what does it mean for aging research?
-
Placentas are a rich source of pluripotent stem cells that could be used for autologous stem-cell transplants.
Rhonda: Dr. Rhonda Patrick here. In this adventure of the "FoundMyFitness" podcast, I'm in Mountain View, California at the SENS Research Foundation. And I have, sitting here with me, Dr. Aubrey Grey in the house. Aubrey is a biomedical gerontologist, and he is founder of the SENS Research Foundation. And as far as I know, the SENS Research Foundation has taken on quite an ambitious goal. And that goal is to help prevent and cure aging. And I think that Aubrey sometimes refers to aging as a disease. And so I'd like to talk a little bit about that. But thank you for being here, Aubrey. And can you please tell us a little bit about the SENS Research Foundation.
Aubrey: Certainly. The SENS Research Foundation is a biomedical research charity. So we're a 501(c)(3), which means taxpayers can get tax benefits if they give us money. And we do research into the diseases and disabilities of old age. And I'm a little bit cautious in using words like cure and disease in relation to aging because we have to remember always that aging is it's a side effect of being alive. It's the consequence of the accumulation in the body of various molecular and cellular changes that are inevitable consequences of what the body does to keep us alive from one day to the next.
Those changes are things that I call damage. And that damage is harmless for a long time because the body is set up to tolerate a certain amount of it. But of course, only a certain amount, which means that eventually, this damage exceeds our tolerance, and we start to decline both mentally and physically, and that's what the diseases and disabilities of old aging are.
So when I talk about cures and about disease, I'm always a little bit careful. I think that the oversimplification that most people make with regard to the difference between diseases on the one hand and aging on the other hand is an extraordinary damaging over simplification, because it makes people unjustifiably over-optimistic about the possibility of curing age-related phenomena that they do think of as diseases, let's say Alzheimer's or cancer, most cancers, or osteoporosis, or whatever, but it makes them also over pessimistic about medical advances to prevent and preempt the aspects of age-related ill health that they don't think of as diseases, like loss of muscle or a decline in function of the immune system, or whatever.
The best way to think about this is that all of these things are part and parcel of the same phenomenon. They are interdependent but nevertheless individual aspects of the accumulation of molecular and cellular damage in the body. And the only way that we're going to bring them under control is by developing a panel of interventions that we can use to periodically repair those various types of damage, and thereby leave the overall abundance of damage in the body below that threshold, such that it's harmless.
Rhonda: Right. So let's dig a little bit more into these types of damage because I talk quite frequently about damage myself. And so typically, when I think of aging, I also think of the degeneration, the accumulation of damage, and degeneration of tissues of cells as a consequence of the accumulation of this damage, and at the same, the inability of our capacity to repair damage, to prevent the damage, also declining. So it's what sort of that this balance, imbalance that begins to have we have more damage accumulating and less capable of repairing the damage as you mentioned.
Aubrey: Okay. So I think I actually would like to stop you there for a moment because a very important thing that an enormous number of even the even gerontologists tend to overlook is that this change in the balance between damage and repair has to be caused by something, right? You do indeed get in old age, a more rapid creation of damage, and a less rapid removal of damage, repair of damage, and thus, you get an accelerated accumulation of the overall amount of damage. But why? The answer is because of the damage that was accumulating early in life, throughout life, even starting before we're born, that we could never repair at all. That is the clock of aging, it's the accumulation of damage that we simply don't have any genetic capability to repair even when we're young.
When that accumulates, it does two things. It accelerates the accumulation, the creation of other damage, and it also impedes everything about the body, including the damage repair mechanisms that we have. So we get less good at repairing the damage that we used to be good at repairing.
Rhonda: Yes, that actually makes perfect sense. The accumulation of this damage is that we do not repair, we simply cannot repair during our youth, will eventually either damage the DNA inside of our cells and that will change the function of certain genes, maybe possibly repair genes or genes that help us deal with this damage. They may change the function of the cell itself so the cell might become more stiff, and that changes the way the cell is functioning, or they may change the way proteins are the function of proteins because now proteins become aggregated and all sorts of changes happen. But in addition to that, it also may lead to epigenetic changes, which also can change the expression and function of genes. And so I think that putting it that way does make sense.
Aubrey: Well, actually, you bring up another important point, especially with your mention of epigenetics, because epigenetics is terribly fashionable within gerontology right now.
Rhonda: Can you explain what epigenetics is for people?
Aubrey: Epigenetics basically is the study of the changes that happen in cells whether as a result of aging or as a result of anything else, that cause differences in which genes are turned on and which genes are turned off. So typically, these will range from things at the DNA level itself, methylation of cytosines for example, up through modifications of histones, these proteins that DNA is wrapped around, up through higher level changes to the packing of chromosomes and to chromatin an awful lot of different things change the behavior with which a cell actually decides which proteins to express and which ones not to. But this...
Rhonda: Let me just interrupt real quick. And for those of you that don't know what express or turn on or off, it just essentially it means, what he's referring to is that when a gene is turned on or if it's expressed, it's active, it's doing the function it's supposed to do. If the gene is turned off or not expressed, that just means that the gene is there but it's not doing its function. It's almost as if it's not there.
Aubrey: But here's the thing. When you see a change late in life, you always have to ask yourself if this change happening as part of aging, or is it happening as an adaptation to part of aging? Is it happening, in other words, to minimize the pathogenic consequences of some other change? And epigenetic changes in aging are pretty much entirely that latter thing, they're adaptations that are good for us, to make the best of a bad job, namely the non-genetic things that are happening elsewhere. We know this simply because they are coordinated. When we look at tissue in bulk, when we look at lots of cells all at the same time and we ask, "What's happening in terms of the gene expression changes?" what we're seeing is a coordinated response. It's got to be coordinated because otherwise, it wouldn't be happening at all in the bulk of cells on average.
So it's bound to be an adaptation, it's genetically programmed. It's happening because the cells know what their environment is, whether intracellular or extracellular, and they're responding to that environment in the same way that they might respond to an infection, or to inflammation, or whatever. The only way you can actually ask questions about epigenetics that are meaningful with regard to actual aging, rather than adaptations to aging, is by looking at individual cells.
Look at individual cells, single-cell analysis, then you can quantify the noise, the amount of variation that's happening without any kind of genetic direction. And we're actually doing that. We have had a project for a few years now in the Albert Einstein College of Medicine in New York looking at precisely this. We're looking specifically at methylation rather than the other aspects of epigenetics. And we're asking, does the epigenetic noise in various tissues increase with age (wouldn’t be much). And the hypothesis that we are pursuing is one that I put forward some time ago now, which is essentially that no, it won't, or at least not to a detectable degree. And the reason it won't is because the quality of DNA maintenance and repair, whether genetic or epigenetic, is driven, in an evolutionary sense, by the need not to die of cancer before you've reproduced.
Cancer is, by far, in my view, the biggest problem of DNA repair and maintenance, because it can kill you with just one cell going seriously wrong in the wrong way. Whereas, anything that doesn't have to do with the cell cycle has to affect an awful lot of, a high proportion of the cells in a given tissue, before it starts to be pathogenic. And that means a lot of cells.
Rhonda: Yeah. So this is all very interesting. Are you familiar with the work that's come out of UCLA from Steve Horvath, I think? So he's shown that from multiple tissues from humans, blood cells, and also different biopsies from different samples, that there's a pattern of methylation that appears to be specific to age. And it's so precise that researchers can look at this methylation pattern from, for example, lymphocytes taken from a person and they can identify the person's age plus or minus four years with 96% accuracy.
Aubrey: Yeah, okay. So let me talk about that actually. Because yes, I know Steve's work pretty well, and I've discussed it with him. And actually, a lot of people have oversimplified what he's been doing and what he's seeing from this. So you're right, there's this extraordinary correlation R-squared of 96% that he's got. He's found this particular set of CpG islands of things that change during age in terms of their methylation that change so uniformly that you get this amazing R-squared.
Now, what does all that actually mean? Well, first of all, you've got to remember that actually if you look in the adult part of life, let's say 23 to 70 or 80, then the R-squared is much lower. It's like 70%, something like that. Second thing you gotta remember is that that's a good thing, because it means you've got some variation to actually work with. If you actually really had something totally linear, right, then first of all, it wouldn't tell you who's aging more quickly and who's aging more slowly. But secondly, it would tell you that your signature is the list of the least important things in aging, the things that are just trundling on in a trajectory that was set during development because evolution hasn't had the faintest motivation to stop them trundling on.
Rhonda: Well, what I found interesting from the research was more the clusters of genes that this was involved around. They were a DNA repair...
Aubrey: Kinda. Kinda. But that's always dangerous. I mean I was actually involved in the very early days of the gene ontology. And I always had doubts about whether it would be misused. And I feel that it is being misused in some ways here. I think that one...Well it's very hard to factor out the multiple hypothesis problem when you're using that kind of analysis of GO terms. Essentially, you've got to ask yourself how many different types of gene, whether it's in terms of function, or process, or whatever, and how many...I mean, what proportion of those genes are affected. It's terribly, terribly easy to run with the first thing you see when you half close your eyes when you look at that kind of data. And I think a lot of people have been doing that. But I'll do what I'm interested in with regard to Horvath's work and related work.
What I'm interested in is when they have looked not at the enormously good R-squared, but at the variations from the R-squared, that's why I said that it's good that the R-squared not so high if you look at adults. The thing there is that, then you can actually ask questions like, "Does the subset of the population that are changing that signature of that group of methylation sites more rapidly than average, do they actually exhibit a greater pathology at a younger age?" Or, loss of function in some other way that you can measure even at a relatively young age. Very recently, just a few weeks ago, there was an extremely interesting paper that came out of a group in New Zealand where they had done exactly this question. They had basically looked at, I can't remember how many people they looked at, but they did a longitudinal study over if I remember rightly 12 years, and there were early adults here. We're talking, I think the ages were something like 26 through 38. And they looked for this kind of variation.
If we can combine that kind of analysis with the kind of methylation analysis that Horvath has developed, then I think we'll be able to ask some very intriguing questions about the predictability of age-related ill health. But now I want to finish my answer by talking about what this means for our work. And this is actually really important because a lot of people overlook this. It's terribly, terribly fascinating that some people age more quickly than others, and some species age more quickly than others, and the whole of gerontology, for more than a century now, has been essentially founded on the idea that if we understand that variation really, really well, we might be able to translate that variation into some kind of therapeutic regimen to turn fast agers into slow agers.
And that I wouldn't object, that would be great. But we've got to remember a couple of things about that approach. Number one, it doesn't work so well if you only apply it late in life, because all it does is slow down the accumulation of damage rather than repairing damage, which is what we're all about. So that's bad enough. We'd like to help people who have the misfortune to be in middle age already or maybe older. The other thing is no one's actually having success in this. Why not? Because metabolism is really complicated. Messing around with this vast network of undocumented spaghetti code that keeps going from one day to the next the idea of stopping it from doing the thing we don't want it to do, the creation of damage, without also stopping it from doing things that we need it to do it's crazy, it's never going to happen. So I really don't think that even if we learn plenty by these methods, that it's going to have all that much impact on the development of actual therapies.
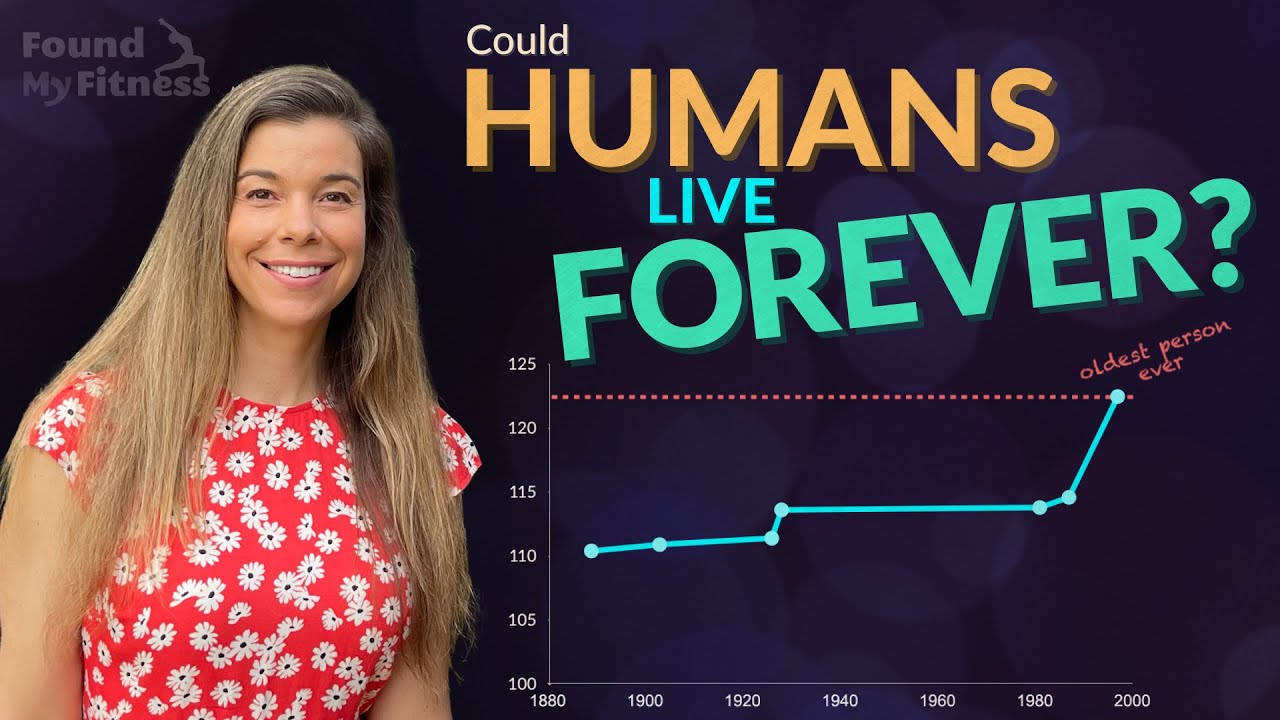
Rhonda: What about some of the more recent methods, for example the CRISPR where this technology where now, I'm going to totally oversimplify this for people. But the ability to specifically target a gene and clip it out, and replace it with another gene or a version of the gene that's more active or less active depending on what it is you want. I think that this new technology for CRISPR, for example, dramatically changes a lot of things because...I mean, even if we look at, for example, centenarians, semi-centenarians, which live to be about 105, or semi-supercentenarians live to be 105, and then the supercentenarians, which are about 110 plus.
A recent study came out from I think it was Tokyo, and Newcastle, I think. I don't know if you're familiar with the study. But essentially what the study did, and it was the largest cohort of the semi-supercentenarians and the supercentenarians. And what they found was that they looked at a variety of different biomarkers. So they looked at inflammatory biomarkers, they looked at lipid profiles, glucose, they looked at immunosenescence. So when your immune cells no longer are living and dividing, they basically sit around, and they're not dead, but they're doing more damage, because they're producing more inflammatory things that are damaging other cells. So it's like spreading more nasty stuff around. They looked at immunosenescence, and then they also looked at telomere lengths.
And then they also looked at like diseases, and then they looked at organ, for like liver function, kidney function. And so, anyways, they're correlating all these factors. And what they found was that inflammation was the only thing that drove aging in all the groups. So inflammation, the higher the inflammation, the higher the risk of death of non-accident, you know? So age-related diseases, cardiovascular disease, cancer. And this was true for all the groups.
But what was really also interesting was that the centenarians, there was a positive correlation between inflammation and immunosenescence, which was essentially lost in the super centenarian group. And I don't know why that is, but the immunosenescence...So essentially the inflammation went up and then the supercentenarians as the inflammation went up, they died. There was a positive correlation. But the immunosenescence seemed to stay around the same for whatever reason. So in my mind, I think, "You know, well, we know that these supercentenarians, that's possibly around a 25% to 30% increase in human lifespan." So human lifespan in the United States is average around 79 years old. If we could live to be 115 and live to be healthy, that's fantastic.
So we know that it can be done with these supercentenarians, and we know that that there's a lot of genetic factors that are playing a role on this, I mean, obviously these people have lower inflammation compared to non-centenarians, they also showed that. And so, and inflammation is upstream of a lot of damage. It's upstream of the damage that's damaging DNA, proteins in the cells, lipids etc., etc. So if we can use CRISPR technology to go in and replace say, give it more anti-inflammatory capabilities, and this has been shown also in mice. I don't know if you've seen this study, but NF-kB, which is a gene that produces a protein that regulates a lot other genes that are pro-inflammation, so they cause inflammation, but it also has an anti-inflammatory component to it.
And when you take away that anti-inflammatory component and put it in mice, what happens is every time there's an immune response, every time inflammation happens, which leads to chronic damage, as you talk about, there's a low level inflammation and it drives aging prematurely in mice.
Aubrey: All right, so. Big question there.
Rhonda: Yes.
Aubrey: Let me just give a fairly big answer. Let me start with a very simple thing. Overtly clear that inflammation is a double-edged sword. That we need it, the reason we have it, as with anything that's genetically carried over that hasn't just mutated into oblivion over evolutionary time, is because it's good for us. Because it's an essential component of how we survive infections. However, there are certain aspects of age-related damage accumulation, which because they are only age-related, are not very interesting to evolution.
And therefore, evolution has not taken the trouble to improve the precision of the inflammatory response so as to discriminate between things that the inflammatory response can actually help with, namely the elimination of infections, and things that the inflammatory response actually exacerbates, namely the accumulation of damage that is not an infection, like oxidized cholesterol, or whatever. So that means that, yes, it's likely that, it's no surprise to us, that when you look at a very, very elite population, the population that live to 105, 110, then they will overwhelmingly have a weak inflammatory response, because that is the only way that they will have been able not to succumb at the age of 80 or 90 to atherosclerosis or Alzheimer's, which are definitely driven partly by the inflammatory response.
However, what also needs to be taken into account is that plenty of people aged 80 and 90 and 100 die of infections. So, yes, these people got genetically lucky because they didn't get an excessive inflammatory response to those age-related problems, but they also got environmentally lucky in that they didn't die of infections. Or maybe they just had a really strong adaptive immune system that compensated for the weak inflammatory response, and so on. So there's a real trade-off here and what this adds up to is that we cannot conclude that it would necessarily be a good idea to take people in their, let's say, 60s or 70s and damp down their inflammatory response.
Rhonda: What about bump up their anti-inflammatory response?
Aubrey: It amounts to the same thing. If we're talking about the strength of the inflammatory response, as opposed to the strength of other aspects of the immune system, like T cells and B cells, then we are engaging in a change of a trade-off. We are giving people less...We are reducing people's risk of rate of...Well, likely rate of progression of atherosclerosis and Alzheimer's and such alike, but we were also increasing their risk of dying pneumonia. Simple as that.
And the best way to deal with this is to find a best of both worlds solution, to let people have the strong inflammatory response that they need in order to be protected well against infections, but to fix the problem of maladaptive activation of the immune response. And what we're kind of doing exactly that, not by changing the inflammatory response itself, but rather by changing the targets. Ultimately, what's happening in atherosclerosis is that the inflammatory response is being activated by the accumulation of indigestible waste products, specifically oxidized cholesterol, in macrophages in the artery wall, which turn to foam cells, and generally make cells around them angry.
If that didn't happen, if we could get rid of that oxidized cholesterol, then it wouldn't matter at all how strong your inflammatory response was. You would not get atherosclerosis. Same for Alzheimer's. Ultimately, Alzheimer's has an inflammatory response because the stuff it's reacting to, it's amyloid and tau and so on. If we can get rid of those materials, stop them from accumulating to an inflammation-triggering level, then we won't get an inflammatory response even in people with a strong inflammatory genetic profile.
Rhonda: Has the SENS Foundation considered using some technologies that are sort of already present in the body? For example, you mentioned Alzheimer's disease, and recently this glymphatic system has been discovered where we now, when we sleep, we know that cerebral spinal fluid squirts up into our brain and literally washes out the amyloid plaques and other buildup of these extracellular aggregates that are in our brain. Has the SENS Foundation thought of any way to use that system somehow?
Aubrey: We're looking at it. We're very interested in all ways of getting rid of molecular garbage, whether the garbage is intracellular or extracellular, and whether the getting rid of is destroying it on site or flushing it into a place where it gets destroyed in other ways we're into all of this thing. We keep our eyes very open and our minds very open with regard to what's going to work.
Rhonda: That's good to know. What about the new...I find this very interesting, the parabiosis where we can take, we, I mean scientists, can take blood from a young animal and transplant it in an old animal, and essentially reverse some biomarkers of aging in multiple organs.
Aubrey: Yeah, it's very exciting. We're actually funding a postdoc at Berkeley doing, working in this area, in one of the top labs in this area. Of course parabiosis itself is not a therapeutic regimen. I mean, presumably, you would not be too keen to do that.
Rhonda: Right.
Aubrey: But it's definitely a great way to make discoveries. And it, of course, leads to alternative versions like plasma exchange and phoresis ways of altering, of taking things out of the old blood, or putting things into the old blood, so as to achieve the same effect that parabiosis would. Of course, in order to do that, you need to know what to take out or put in. And a lot of the problems that parabiosis research faces at the moment is that that's really laborious and tricky to find out in any kind of systematic way. The few hits that people have had so far in terms of factors that seem to actually have some kind of causal role like GDF11...And so these things we found more or less serendipitously. And everyone knows that there's likely to be a number of others out there, perhaps very likely to be ones that are more central to the effect, but which have not been found just because they're a little more counterintuitive.
Rhonda: So I envision if these factors can be identified, GDF11, growth differentiation factor 11, was thought to be possibly playing a role in causing muscle stem cells to divide and proliferate, and possibly in the brain as well. But others have not been able to confirm that, but if...
Aubrey: Yeah, watch this space. That's going to run and run.
Rhonda: Yeah. I mean, I'm not sure. I mean, all I know is that I'm excited about the research in general. And whatever the factors are, I envision possibly making recombinant proteins. I mean, people are using EPO, erythropoietin. I mean human growth factor. So some the same sort of deal.
Aubrey: The big thing that needs to be taken into account here is that the factors that change in their abundance in the blood during age, whether up or down, and that may have an effect in terms of if you like transmitting pathogenic damage from one place in the body to another, those things are, first of all, they're not necessarily just proteins. We also have to worry about cells, the fact that you have changes in the relative abundance of different types of T cells for example. And there's also a small molecule, glutathione, things like that. You know, things that may simply not be amenable to rejuvenation by measures like plasma exchange, but only by, from that parabiosis. Then we would have to look at a different model.
But I want to also emphasize something I just alluded to a moment ago, which is that we are talking here not about mechanisms whereby damage is created. We are talking about mechanisms whereby damage is transmitted from one part of the body to another. After all, that's what the circulation is, it just takes stuff from one place to another. It doesn't create damage. The damage comes from somewhere. So we always need to be looking out for the possibility that we can find the cells that are the source of the changes in the blood, and change those cells back to a rejuvenated state, and by that means, rejuvenate the blood.
Rhonda: Well, I think that the source of damage, in the sense of the circulation and the blood cells the immune cells in the blood, are just that. You know, when you activate macrophages, they dump out hydrogen peroxide and all sorts of reactive nitrogen species.
Aubrey: Yeah. That's not really what I'm talking about. I mean, I'm talking about, for example, well, let's take the thymus for example. The thymus itself shrinks, as we were saying earlier, and that is largely responsible for the fact that in an older person, there are fewer naive T cells and more memory T cells. You know, if we can fix the thymus, then we fix that problem. And, yeah, I can make many other example.
Rhonda: Yeah, that really makes sense. Something else talking about this damage and you and I both agree that...I mean, I think the accumulation of damage, intracellular, so inside the cell, outside of cells, on cell membranes, proteins, DNA, on and on. I mean, I think that is a driver of aging and essentially causes aging.
Aubrey: I would say is aging.
Rhonda: Yeah. And I focus more on an easier solution, which is the nutritional aspects of preventing, or of allowing your body to metabolize and produce energy the best it can. And even though you're still going to age, like even if you're at the optimal amount, I mean, even if you have the optimum amount of micronutrients, you have these minerals and vitamins that are essential to run your metabolism, they're essential to run enzymes that repair damage so on. The fact is you will still age. The question is how much better will you age?
Aubrey: So, I should put... Yeah, exactly. The question is how much better? And I'm all for all of that work, you know. A large part of why Bruce and I became friends long time ago was because I absolutely endorse the idea that we need to do the best we can for the population to get them to an average level of nutrition. But I think the critical thing to understand is average. If you ask about the difference in terms of health expectancy and life expectancy between, let's say, the middle 10% of the population and the bottom 10%, it's a large difference. And that's the difference that we're talking about here, the difference that Bruce is trying to do something about by making sure that if the poor won't eat fruit, then that we find a multivitamin and so on.
But if you look at the opposite end of the spectrum, if you look at the difference between the middle 10% and the top 10% in terms of health expectancy and life expectancy, it's basically nil. Of course, I'm factoring out genetics here, I'm talking about lifestyle. I'm talking about things that we can modify. And that's really important to remember, because it's so easy from the popular press and so on to get the impression that if you just do what your mother told you to and do it really well you eat a really good diet, and you get a lot of exercise, and you never drink anything, you never smoke, and so on, then you're actually going to live 20 years longer than you otherwise would. When in fact, the message of all of the data we have, epidemiological or anything, is that it's probably closer to two years, if that. You know, I mean other example I like to give is just a very simple one looking at national life expectancies.
So people laugh at the USA a great deal because of the fact that it sits at number something like 45 in the league table of longevity among the industrialized world, despite the fact that you guys spend far more per head on medical care than anybody else. But if you look at the absolute numbers and you look at the actual difference in number of years in life expectancy between the USA and the number one big country, namely Japan, it's only four years, only four years.
Rhonda: Yeah. No, I'm familiar with this data. It's five years actually. The average lifespan experienced in the United States is 79, and Japan, it's 84. And, what's really interesting...So first of all, I'm not sure that Japan has the optimal diet in general, they they're getting all the micronutrients. I mean there's a lot of factors here. But what I do find interesting is if you look at the data...So right now, the average difference is five years. And in between males, it's four years, between females it's six years. If you look at the data from 2012, 2013, Japan has gone down in their life expectancy. So their average life expectancy has gone down by a year. U.S. has gone down by a year. So there was a bigger difference, it was six years. But what's really interesting is that the male life expectancy in Japan has gone down by almost four years, three points or so.
Aubrey: Okay. So those are the kinds of fluctuation in data that I find very untrustworthy.
Rhonda: Yeah. I mean, I think that it's possible that Japan's becoming more westernized, males also smoke like chimneys over there. So I mean, there's a lot of other factors that come into play.
Aubrey: Okay, that could be quality of data, you know...
Rhonda: Could be quality of data, exactly. I think that if...Like you mentioned earlier if you really want to look at the effect of diet on lifestyle, then looking at obesity. Obesity is associated with a seven-year reduction in lifespan. Morbid obesity is associated with a 14-year reduction.
Aubrey: Oh, don't get me wrong, of course. But that's the low end versus middle that I was talking about.
Rhonda: But it's growing problem in the United States. You know, obesity is...
Aubrey: Well aware of that. Jay Olshansky of course has been very prominent in publicizing this problem and predicting that unless we do something very serious about basically epidemic, then we are in danger of seeing a fall in the life expectancy in the USA. But of course, we haven't seen that yet because the problem is too new.
Rhonda: It's what?
Aubrey: The problem is too new.
Rhonda: Too new, yeah. So I do think that I talk a lot about what role micronutrients have in diet, and metabolism, I mean, B vitamins are running your mitochondria, magnesium's needed for gene repair enzymes, vitamin K is needed to coagulate, blood coagulation, on and on. So it also plays a very important role. And I do think it absolutely affects the way you age, especially if you're talking about living in an unhealthy eating refined carbohydrate sort of diet versus eating your greens, and exercise, and things like that. But even with that said, and doing all those things, you're still going to age, because you can't stop the breathing in oxygen and eating food, this process that is coupled together to make energy. Well, it's, inherently makes damage, yeah. And there's no stopping it. I mean, no matter what amount of nutrients you get.
Aubrey: Which of course is exactly where I came in, back in 2000, with the realization that even though we couldn't stop this damage from being created, we could go in and comprehensively, not necessarily 100%, but very, very comprehensively, repair that damage, and thereby, keep its overall level of abundance to a level that the body is set up to tolerate with full function.
Rhonda: So with these discoveries, CRISPR technology pluripotent stem cells...
Aubrey: These are huge things.
Rhonda: Is this advancing your research?
Aubrey: Absolutely, absolutely. It's advancing our research just as it's advancing everybody else's. These are techniques, technological innovations that, just like the fact that we can now have the sequence of a human genome, they just make things easier and faster. CRISPR I would single out as a particularly important advance, because there are definitely quite a few things we're going to have to do in getting this damage repair to be comprehensive that involve genetic modifications. And some of those genetic modifications are going to be possible to do ex vivo, in stem cells that we then reinject into the same person. Some of them are not. Some of them are just going to have to be done by bona fide somatic gene therapy.
And as we know, somatic gene therapy has had a rocky ride over the past 20 odd years, because it's really difficult to make it safe. And the fundamental reason it's so difficult to make it safe is because the viruses, the vectors that are used to get engineered DNA into places are not easy to control in terms of where they insert themselves into the DNA. And thereby, they are not easy to control in terms of what damage they may do, making a cell cancerous, etc.
CRISPR on the other hand, started out being pretty good at its site specificity. And better than that, as time's gone on, very rapid advances have been made such that now, it's just out of site site-specific. It's incredibly high fidelity. That means that one can increase the titer, the amount of engineered DNA that you stick into their body that's supposed to go and modify cells. And by increasing the titer, you can increase the penetrance, the proportion of cells that are actually modified in the way you want, without increasing the off-target effects, because the off-target effects are being eliminated by the nature of CRISPR.
Rhonda: Yeah. I think that using CRISPR, I think there's obviously a lot of things that need to be overcome, like getting it to the right tissues. I mean, you still have to have some sort of targeting sequence to say, "Okay, we want CRISPR." It's easier to do ex vivo when you take your blood cells. I think...
Aubrey: Ex vivo is always going to be easier.
Rhonda: Right. But saying we want to get this to the liver, or we want to get this to the heart.
Aubrey: On muscle, yeah. That's right.
Rhonda: Or muscle. Yeah, much more difficult. And some of these technologies that you were describing, about engineering cells to have certain viruses that make them go somewhere or change a gene, also we don't know what their effects are in terms of putting them in our body. Are they gonna cause cancer. I see sort of the same challenges with the induced pluripotent stem cells. So being able to make tissue...for example, take a skin cell from your body and give it the right genetic combination to trick it and reprogram it into becoming a stem cell, a pluripotent stem cell, so that it can form any cell in the body, that also is takes some viruses at this point, I think...
Aubrey: Well, first of all, no, there are plenty of ways now that have been perfected that induce pluripotency without actually using viruses at all. The most recent one that got a lot of attention a month or two ago was when Helen Blau's group at Stanford showed that they could do it with messenger RNA. But it's also been done just by electroporating proteins in.
Of course, the problem here is that the actual efficiency is rather low in many cases. But that's improving. The other thing is the quality of the reprogramming. So the original Yamanaka factors, they work pretty well. But Jean-Marc Lemaitre in France a few years ago showed that if you use six factors, then you can get a much more high fidelity reprogramming, you can even reprogram senescent cells, which you couldn't do with the regular Yamanaka factors, and so on. You know, these are things are...you know, it's an enormous field, and it's progressing really fast, simply because they can. And I'm overjoyed it's going to make a lot of things easier.
Rhonda: That's really exciting. Are you familiar with the fact that placenta is a good source of pluripotent stem cells?
Aubrey: Doesn't surprise me the slightest.
Rhonda: Yeah. And that it's just being trashed every day.
Aubrey: But the point is, of course we want to treat people who are already in middle age, right? So they don't have their placentas any more than they have their umbilical cord or whatever. And if we can do the reprogramming well, then that's fine.
Rhonda: Well, if you have enough placentas being banked, sort of like blood, then you can potentially find a match.
Aubrey: Oh, well, of course. Now, we're talking about falling short of true autologous administration. As far as I'm concerned yes, it's good to have matches that cut out some of the immune, the reaction response because of MHC compatibility. But the fact is, the real McCoy is taking cells from the prospective recipient, reprogramming them, doing whatever you want, and putting them back into the same person. And the only reason that that at the moment is not what everyone's looking at is because it costs a lot of money to do that on a personalized basis. But as time goes on, and we get better and better at these things, that cost is going to reduce, and all this banking stuff is going to be obsolete.
Rhonda: Do you know if the reprogramming of a skin cell into, say a pluripotent stem cell, do you know if it's been shown that everything gets reprogrammed, the epigenetics, I mean, because you're essentially talking about if you take it from an adult who's 50 years old, it's a 50-year-old skin cell, I mean...
Aubrey: So this is what I was saying...Well, this relates to what I was saying a moment ago about using the original Yamanaka technique versus refinements of it. So for sure, it's been shown by a number of groups that the standard methods of creating induced pluripotent stem cells do not 100% erase the epigenetic state that the cell came from. There is a retention of some "epigenetic memory," as people are calling it. That epigenetic memory is considerably less in this system where you use six factors, that I mentioned. And of course, other people are looking at other ways to eliminate it even further. Then again, of course, you've got to ask how much elimination is needed for a particular person.
Is it in fact fine for cells that started out being skin, but you're going to use them for blood to actually have a little bit of skin behavior in them? You know, does it actually matter? You know, these are the questions that people are asking all the time all over the world right now.
Rhonda: Yeah. Well, I'm less worried about that and more worried about the fact that you may now have certain genes that should be more highly expressed at a younger age, to make it younger, not...So for example the cell cycle regulator, ARF p16(INK4a). During early youth, early development, as we're younger, it's silenced, epigenetically silenced. The reason for that is because if it's not silenced, stem cells stop dividing, it essentially says, "stop." And so you want the stem cell to divide.
Aubrey: So there's two answers to that. The first answer is that this is reprogramming, right? So if you're erasing the whole of the epigenetic state of a cell, and taking it back to how it was in the embryo, then you're going to re-differentiate it in the direction you want to the extent that you want. And that's going to make it into let's say, an oligopotent skin stem cell, with its p16 suppressed, the way a regular oligopotent stem cell would be, and the way that you make sure that's true is just by knowing what to do in the re-differentiation process.
So my friend and colleague, Mike West, at BioTime, has been working on this for a while. And that's the main thing that BioTime is really good at, this method that controls and systematizes the redifferentiation process. The other thing to mention though, is that, yes, if you take a bunch of skin cells from an older person, then there's going to be a spectrum of level of expression of, let's say, P16. Now, it may be that the process of dedifferentiation, getting it back to the iPS state in the first place, is actually going to be affected by that, such that the cells that actually give rise to your iPS cells will be preferentially the ones that happen to have low P16 in the sample that you took from the original person. So what, really.
Rhonda: Yeah. So you had brought up this idea a little earlier of kind of at least I think it's somewhat of an antagonistic pleiotropy, when you're talking about for example the immune system. You know, it's sort of, you want an active immune system because you want to survive through reproduction, you want to not die from some bad nasty infection, but also, this inflammatory process as you get older can accelerate aging.
Aubrey: Yup. So you got to be very careful with antagonistic pleiotropy. It's a overused term. I'm not even sure that one should call the inflammatory response an example of antagonistic pleiotropy. Because remember, the situation in an older individual includes the fact that the rest of the immune system has declined, so that you kinda need a high inflammatory response just to fight off infections. And maybe it's a good tradeoff even in the elderly, irrespective of the fact that it was a different tradeoff earlier in life.
Rhonda: Yeah, I think that's less of an example. More of a better example would be something like growth hormone, or IGF-1, which is very important for development, for growth, it causes muscles to repair damage, it actually grows new neurons. I mean, it's a great growth factor. But, as you get older and you have more cells that have accumulated damage, that have more damaged DNA having too much IGF-1 around allows these cells instead of to die to grow. So basically...
Aubrey: Well, so actually, I would say that that's an example rather similar to the inflammatory one, in the sense that, rather being strictly speaking antagonistic pleiotropy, good in the young, bad in the old, this is a case where it's good in the old and bad in the old in different ways. So you want more growth hormone in order to have better muscle, you want less growth hormone in order to have less cancer.
A neurodegenerative disorder characterized by progressive memory loss, spatial disorientation, cognitive dysfunction, and behavioral changes. The pathological hallmarks of Alzheimer's disease include amyloid-beta plaques, tau tangles, and reduced brain glucose uptake. Most cases of Alzheimer's disease do not run in families and are described as "sporadic." The primary risk factor for sporadic Alzheimer's disease is aging, with prevalence roughly doubling every five years after age 65. Roughly one-third of people aged 85 and older have Alzheimer's. The major genetic risk factor for Alzheimer's is a variant in the apolipoprotein E (APOE) gene called APOE4.
A toxic 42 amino acid peptide that aggregates and forms plaques in the brain with age. Amyloid-beta is associated with Alzheimer's disease, a progressive neurodegenerative disease that can occur in middle or old age and is the most common cause of dementia. Heat shock proteins have been shown to inhibit the early aggregation of amyloid beta 42 and reduce amyloid beta plaque toxicity [1].
A concept from evolutionary biology that suggests certain genes may influence fitness differently throughout the life cycle. Genes that exhibit antagonistic pleiotropy increase the odds of successful reproduction early in life but have deleterious effects later in life. For example, mutations causing overproduction of sex hormones may increase the sex drive and reproductive success but could, hypothetically, promote prostate cancer (in males) and ovarian cancer (in females) with aging.
Tumor suppressor gene, the focus of which is found to be deleted or silenced in many kinds of tumors. For example, of the 100 primary breast carcinomas, approximately 41% have p14ARF defects.
A disease characterized by the deposition of fatty plaques on the inner walls of arteries. Something is said to be atherogenic when it promotes the formation of fatty plaques in the arteries. Atherosclerosis causes coronary artery disease.
Involving one person as both donor and recipient.
A measurable substance in an organism that is indicative of some phenomenon such as disease, infection, or environmental exposure.
A person who is 100 or more years old.
A waxy lipid produced primarily in the liver and intestines. Cholesterol can be synthesized endogenously and is present in all the body's cells, where it participates in many physiological functions, including fat metabolism, hormone production, vitamin D synthesis, and cell membrane integrity. Dietary sources of cholesterol include egg yolks, meat, and cheese.
A tightly coiled molecule of DNA found in the nucleus of a cell. Chromosomes contain the genes and other genetic material for an organism. Humans have 46 chromosomes arranged in 23 pairs. Each chromosome is comprised of long stretches of DNA wrapped around proteins called histones, which provide structural support. At the end of each chromosome is a repetitive nucleotide sequence called a telomere. Telomeres form a protective “cap” – a sort of disposable buffer that gradually shortens with age – that prevents chromosomes from losing genes or sticking to other chromosomes during cell division.
Short stretches of DNA where the frequency of the cytosine-guanine (CG) nucleotide sequence is higher than in other regions. (The "p" in CpG reflects the presence of a phosphate group between the two nucleotides.) DNA methylation commonly occurs on CpG islands. If it occurs on a CpG island in the promoter region of a gene, it turns off, or "silences," the gene's expression.
A gene-editing technology that employs a prokaryotic enzyme called Cas9 and an RNA guide molecule to home in on its target DNA. Cas9 then edits the DNA to disrupt genes or insert desired sequences. Cas9 makes up part of a prokaryotic immune system, which confers resistance to foreign genetic elements such as plasmids and phages.
The biological process in which a cell matures and specializes. Differentiation is essential for the development, growth, reproduction, and lifespan of multicellular organisms. Differentiated cells can only express genes that characterize a certain type of cell, such as a liver cell, for example.
A major contributing factor to aging, cellular senescence, and the development of cancer. Byproducts of both mitochondrial energy production and immune activity are major sources of DNA damage. Additionally, environmental stressors can increase this base level of damage. DNA damage can be mitigated by cellular repair processes; however, the effectiveness of these processes may be influenced by the availability of dietary minerals, such as magnesium, and other dietary components, which are needed for proper function of repair enzymes.
Any of a group of complex proteins or conjugated proteins that are produced by living cells and act as catalyst in specific biochemical reactions.
Genetic control elicited by factors other than modification of the genetic code found in the sequence of DNA. Epigenetic changes determine which genes are being expressed, which in turn may influence disease risk. Some epigenetic changes are heritable.
A hormone secreted by the kidneys that increases the rate of production of red blood cells in response to falling levels of oxygen in the tissues. EPO is also involved in the wound healing process, and plays a role in the brain's response to neuronal injury as well.
Taking place outside an organism. Ex vivo conditions allow experimentation of an organisms cells or tissue under more controlled conditions than is possible in in vivo experiments (in the intact organism).
Fat-laden macrophages seen in atherosclerosis that are formed when the body sends macrophages to the location of a fatty deposit on the blood vessel walls.
The process in which information stored in DNA is converted into instructions for making proteins or other molecules. Gene expression is highly regulated. It allows a cell to respond to factors in its environment and involves two processes: transcription and translation. Gene expression can be turned on or off, or it can simply be increased or decreased.
The scientific study of old age, the process of aging, and the particular problems of old people.
An antioxidant compound produced by the body’s cells. Glutathione helps prevent damage from oxidative stress caused by the production of reactive oxygen species.
A system that clears the brain of metabolites and other waste. The glymphatic system comprises a vast arrangement of interstitial fluid-filled cavities surrounding the small blood vessels that serve the brain. During sleep, these perivascular structures increase in size by more than 60 percent. This allows a “flushing” operation in which waste products can be eliminated. The glymphatic system also facilitates the distribution of essential nutrients such as glucose, lipids, and amino acids, as well as other substances, such as growth factors and neuromodulators.
A naturally occurring substance capable of stimulating cellular growth, proliferation, healing, and differentiation. Growth factors typically act as signaling molecules between cells. Examples include cytokines and hormones that bind to specific receptors on the surface of their target cells.
The chief protein components of chromatin found in eukaryotic cell nuclei that package and order the DNA into structural units called nucleosomes acting as spools around which DNA winds, and playing a role in gene regulation.
The gradual deterioration of the immune system brought on by natural age advancement. Immunosenescence is considered the most important reason for the increased rate of infections (and cancers) in older adults and is believed to be the diminished or exhausted function of the immune system that naturally occurs with aging.
A critical element of the body’s immune response. Inflammation occurs when the body is exposed to harmful stimuli, such as pathogens, damaged cells, or irritants. It is a protective response that involves immune cells, cell-signaling proteins, and pro-inflammatory factors. Acute inflammation occurs after minor injuries or infections and is characterized by local redness, swelling, or fever. Chronic inflammation occurs on the cellular level in response to toxins or other stressors and is often “invisible.” It plays a key role in the development of many chronic diseases, including cancer, cardiovascular disease, and diabetes.
A type of white blood cell. Macrophages engulf and digest cellular debris, foreign substances, microbes, cancer cells, and oxidized LDL in a process called phagocytosis. After phagocytizing oxidized LDL, macrophages are referred to as foam cells.
A form of RNA, transcribed from a single strand of DNA, that carries genetic information required for protein synthesis from DNA to the ribosomes.
The thousands of biochemical processes that run all of the various cellular processes that produce energy. Since energy generation is so fundamental to all other processes, in some cases the word metabolism may refer more broadly to the sum of all chemical reactions in the cell.
A biochemical process involving the addition or subtraction of a methyl group (CH3) to another chemical group. In epigenetics, a methyl group is added to an amino acid in a histone tail on DNA, altering the activity of the DNA segment without changing its sequence. Under- and over-methylation are referred to as hypomethylation and hypermethylation, respectively.
Vitamins and minerals that are required by organisms throughout life in small quantities to orchestrate a range of physiological functions. The term micronutrients encompasses vitamins, minerals, essential amino acids, essential fatty acids.
Tiny organelles inside cells that produce energy in the presence of oxygen. Mitochondria are referred to as the "powerhouses of the cell" because of their role in the production of ATP (adenosine triphosphate). Mitochondria are continuously undergoing a process of self-renewal known as mitophagy in order to repair damage that occurs during their energy-generating activities.
A rapid-acting transcription factor that responds to harmful cellular stimuli, such as reactive oxygen species, IL-1B, bacterial endotoxin (lipopolysaccharide or "LPS"), ionizing radiation, and oxidized LDL. Incorrect regulation of NF-kB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. Several viruses, including the AIDS virus HIV, have binding sites for NF-kB. In the case of HIV, the presence of NF-kB is believed to be involved in switching the virus from a latent to an active state.
The ability of progenitor cells to differentiate into a few cell types. It is a degree of potency that is less than that of multi-, pluri-, or totipotent, but greater than that of unipotent. Examples include lymphoid or myeloid stem cells.
In general, anything that can produce disease. Typically, the term is used to describe an infectious agent such as a virus, bacterium, prion, fungus, or other microorganism.
Capable of developing into any type of cell or tissue except those that form a placenta or embryo.
A portmanteau of the words protein and homeostasis. Proteostasis is maintained through the competing and integrated biological pathways within cells that control the biogenesis, folding, trafficking and degradation of proteins present within and outside the cell. Proteostasis deteriorates with age. As a result, the prevalence of age-related protein misfolding diseases, such as Alzheimer’s disease and Parkinson’s disease, increases.
Senescence is a response to stress in which damaged cells suspend normal growth and metabolism. While senescence is vital for embryonic development, wound healing, and cancer immunity, accumulation of senescent cells causes increases inflammation and participates in the phenotype of aging.
A cell that has the potential to develop into different types of cells in the body. Stem cells are undifferentiated, so they cannot do specific functions in the body. Instead, they have the potential to become specialized cells, such as muscle cells, blood cells, and brain cells. As such, they serve as a repair system for the body. Stem cells can divide and renew themselves over a long time. In 2006, scientists reverted somatic cells into stem cells by introducing Oct4, Sox2, Klf4, and cMyc (OSKM), known as Yamanaka factors.[1]
- ^ Takahashi, Kazutoshi; Yamanaka, Shinya (2006). Induction Of Pluripotent Stem Cells From Mouse Embryonic And Adult Fibroblast Cultures By Defined Factors Cell 126, 4.
A person who is 110 years old or more.
A microtubule-bound protein that forms the neurofibrillary "tau tangles" associated with Alzheimer's disease. Tau tangles disrupt transport of metabolites, lipids, and mitochondria across a neuron to the synapse where neurotransmission occurs. Diminished slow-wave sleep is associated with higher levels of tau in the brain. Elevated tau is a sign of Alzheimer's disease and has been linked to cognitive decline.
Distinctive structures comprised of short, repetitive sequences of DNA located on the ends of chromosomes. Telomeres form a protective “cap” – a sort of disposable buffer that gradually shortens with age – that prevents chromosomes from losing genes or sticking to other chromosomes during cell division. When the telomeres on a cell’s chromosomes get too short, the chromosome reaches a “critical length,” and the cell stops dividing (senescence) or dies (apoptosis). Telomeres are replenished by the enzyme telomerase, a reverse transcriptase.
A fat-soluble vitamin. Vitamin A is a collective term that includes retinol, retinal, and retinyl esters. Vitamin A plays key roles in immune function, reproduction, and cellular communication. It is best known for its role in supporting eye health and the processes involved in vision. Vitamin A also supports cell growth and differentiation and participates in the normal formation and maintenance of the heart, lungs, kidneys, and other organs. Dietary sources of vitamin A include liver, fish oils, milk, eggs, leafy green vegetables, orange and yellow vegetables, tomato products, fruits, and some vegetable oils.
A type of fat-soluble vitamin. Vitamin K is critical for blood clotting, bone metabolism, prevention of blood vessel mineralization, and regulation of various cellular functions. Naturally occurring forms of vitamin K include phylloquinone (vitamin K1) and a family of molecules called menaquinones (vitamin K2). Vitamin K1 is synthesized by plants and is the major form in the diet. Vitamin K2 molecules are synthesized by the gut microbiota and found in fermented foods and some animal products (especially liver). The body has limited vitamin K storage capacity, so the body recycles it in a vitamin K redox cycle and reuses it multiple times.
A type of fat-soluble vitamin that participates in blood clotting and bone metabolism. Naturally occurring forms of vitamin K include phylloquinone (vitamin K1) and a family of molecules called menaquinones (vitamin K2). Vitamin K1 is synthesized by plants and is the major form present in the diet. Vitamin K2 molecules are synthesized by the gut microbiota and found in fermented foods and some animal products (especially liver). The body has limited vitamin K storage capacity, so the body recycles it in a vitamin K redox cycle and reuses it multiple times.
A type of fat-soluble vitamins that participate in blood clotting and bone metabolism. Naturally occurring forms of vitamin K include phylloquinone (vitamin K1) and a family of molecules called menaquinones (vitamin K2). Vitamin K1 is synthesized by plants and is the major form in the diet. Vitamin K2 molecules are synthesized by the gut microbiota and found in fermented foods and some animal products (especially liver). The body has limited vitamin K storage capacity, so the body recycles it in a vitamin K redox cycle and reuses it multiple times.
Proteins that can reprogram differentiated (mature) cells into pluripotent stem cells. Yamanaka factors are highly expressed in embryonic stem cells in mice and humans. Five Yamanaka factors have been identified: Oct4, Sox2, cMyc, Klf4, and NKX3-1. In a mouse model of premature aging, short-term expression of Oct4, Sox2, Klf4, and c-Myc ameliorated cellular and physiological hallmarks of aging and prolonged lifespan.[1]
- ^ Ocampo, Alejandro; Martinez-Redondo, Paloma; Platero-Luengo, Aida; Hatanaka, Fumiyuki; Hishida, Tomoaki; Lam, David, et al. (2016). In Vivo Amelioration Of Age-Associated Hallmarks By Partial Reprogramming Cell 167, 7.
Hear new content from Rhonda on The Aliquot, our member's only podcast

Listen in on our regularly curated interview segments called "Aliquots" released every week on our premium podcast The Aliquot. Aliquots come in two flavors: features and mashups.
- Hours of deep dive on topics like fasting, sauna, child development surfaced from our enormous collection of members-only Q&A episodes.
- Important conversational highlights from our interviews with extra commentary and value. Short but salient.
Aging News
- The rise of early-onset cancer may be linked to changes in biological aging.
- An epigenetic pace-of-aging test was more strongly linked to risk of death than other aging biomarkers.
- Weekly rapamycin did not enhance exercise gains in older adults and may have limited overall functional improvements.
- Garlic-derived compounds may extend lifespan and support healthy aging by improving metabolism.
- Cardiovascular disease risk remains similar in men and women until the mid-30s, then diverges sharply as coronary disease appears earlier in men.